Why Safe As is not Good Enough
June 14, 2019
Blog

It is likely that patients in the U.S. are benefitting less than they should from technological advances in medical care.
It is likely that patients in the U.S. are benefitting less than they should from technological advances in medical care.
From 1976 onwards, all medical devices under the jurisdiction of the FDA have been assigned to Class I, II, or III, where Class III devices demand the highest levels of regulation necessary to provide reasonable assurance of their safety and effectiveness. In theory, any new device introduced since then is automatically in Class III. However, the FDA’s 510(k) “clearance pathway” allows such a device to be reclassified as Class I or Class II as long as it is seen to be as safe and effective as a “predicate device” already in the market – that is, a piece of pre-existing, legally marketed equipment.
In their announcement of November 27, 2018, the FDA registered their concern that “approximately 20 percent of 510(k)s that were cleared were based on substantial equivalence to a predicate device relied on a predicate that was more than 10 years old” and argued that predicate modernization is a key component of promoting innovation. Although such a new device does not need to be identical to its predicate to establish this “Substantial Equivalence,” it does imply strong similarity with respect to intended use, design, energy used or delivered, materials, performance, safety, effectiveness, labelling, biocompatibility, standards, and other applicable characteristics.
In other words, if a medical device designer comes up with a bold new way of helping people, their new device will be more costly and difficult to bring to market that one which may be less capable, but more similar to something that was introduced 10 years ago.
NOW Technologies’ Gyroset Glory headset, for example, has been designed and developed with the European market in mind (sidebar). When groundbreaking devices such as this hit the U.S. market, there is clearly a rock-solid case for ensuring that it should be exposed to the most rigorous process standards imaginable.
NOW Technologies Gyroset Glory
Traditionally, people with disabilities have to deal with a variety of controllers to interact with their computers, mobile phones, and other devices, including wheelchairs. NOW Technologies created Gyroset Glory to be both a universal controller and a discrete, comfortable headset.
Future versions of the controllers are planned to give wheelchairs an autonomous capability, making them even more groundbreaking.
The GyroSet does not require certification and approval in NOW Technologies’ chosen markets. However, the rigor that results from adhering to such standards appealed to the company, which reasoned that it would ensure the reliability of the product and the safety of its users, and help to minimize future maintenance and warranty costs.

Setting the Standards
NOW Technologies have not been required to adhere to demanding standards. But what if they were? A multi-purpose device like the Gyroset Glory with varying roles to fill may be required to meet a host of them:
- Medical equipment—In some use cases, the product might be viewed as medical equipment. Compliance with IEC60601-1 has become a requirement for the commercialisation of electrical medical equipment in many countries.
- Wheelchair controller—In its more specific deployment as a wheelchair controller, ISO 7176 becomes relevant. ISO 7176-14:2008 specifies requirements and associated test methods for the power and control systems of electrically powered wheelchairs and scooters.
- Autonomous wheelchair—Controllers giving wheelchairs an autonomous capability may require certification to safety-critical standards, such as IEC 62304:2006 "Medical device software - Software life cycle processes."
Most relevant here is IEC 62304 Edition 1.1. It defines Classes A, B and C, which are approximately analogous to the FDA’s Minor, Moderate and Major “levels of concern”. However, the allocation of IEC 62304 classes is arrived at and based purely on the basis of a risk assessment and the standard has no concept of predicates.
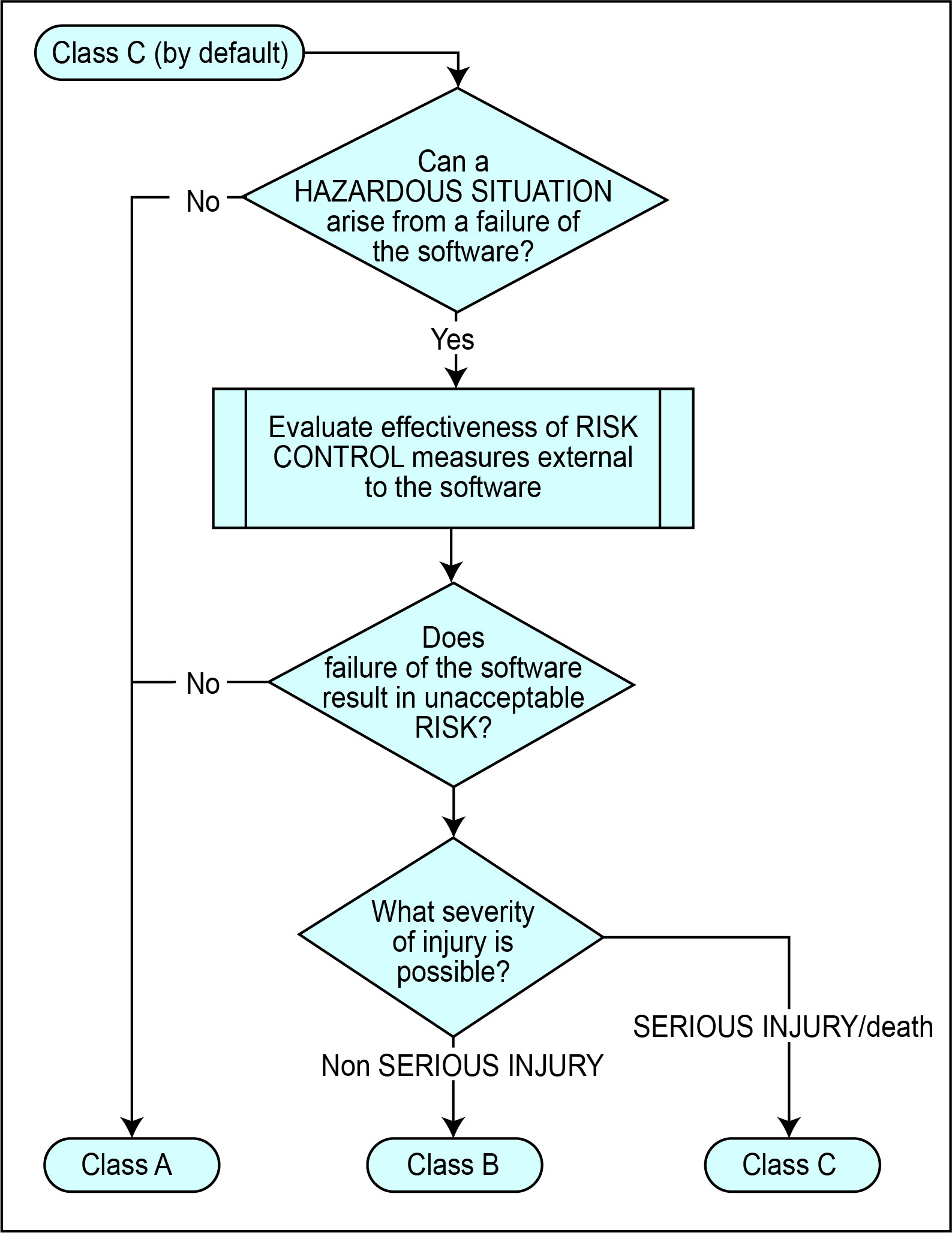
The rigor of the development process for a product developed in accordance with IEC 62304 is therefore reflected in its classification much as for FDA classes, but it is not reduced just because there have previously been similar devices available. If the risk to the patient or user of a serious injury from a malfunctioning device is high, then the rigor imposed by IEC 62304 is equally demanding whether that device represents an innovation, or a repackaging of existing approaches.
Of course, groundbreaking technology is inherently more difficult and more costly to develop, and there will always be manufacturers who stick with established principles for that reason. But there seems no logical argument as to why that line of thinking should be reinforced by permitting derivative products to be developed to a lesser standard than innovative ones.
Shifting the Problem?
If the opportunity exists to reference a predicate design and therefore reduce costs, the temptation not to be so innovative but instead to create a “me too” product must be persuasive. It presents a competitive advantage to manufacturers of derivative devices over innovative ones.
It is clear from the press announcement of December last year that the FDA recognizes that there is a problem. But is the modernization of the 510(k) process to “promote reliance on more up-to-date predicates” actually addressing the core problem, or merely disguising it by ensuring that predicates (and hence their derivative designs) represent more current technology than has previously been the case?
The development processes advocated by IEC 62304 assess the level of risk to the patient entirely on the basis of a detailed assessment of the consequences of failure, with no concept of predicates or Substantial Equivalence. It demands the same development standards for new technology as for established technology. And although it may be easier to achieve those standards with established techniques, that is a different matter entirely to lowering them.